
Recent Advances in the Molecular Visualization and Processing with VTK and ParaView
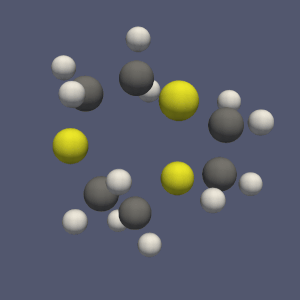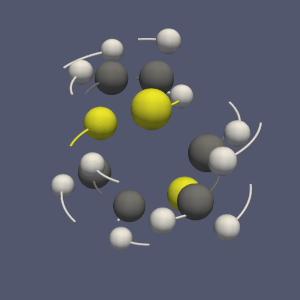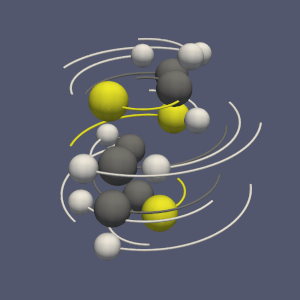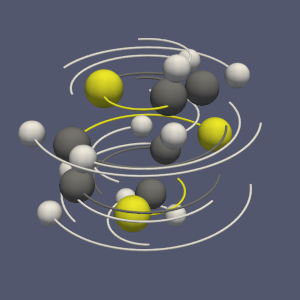
The molecule framework in VTK and ParaView has recently been improved. In this blog we quickly introduce the new features.
Note that the features presented below are available in ParaView 5.6 and VTK master branch (thus in next release VTK 8.2).
Conversion Between vtkMolecule and vtkPointSet
Two new filters enable converting vtkMolecule into vtkPolyData (mesh data structures for 0, 1 and 2D primitives) and reversely:
- “Convert into polydata” (vtkMoleculeToPolyDataFilter)
- “Convert into molecule” (vtkPointSetToMoleculeFilter)
These conversions are based on an atom-point mapping (1 atom = 1 point) and on a bond-line mapping (1 bond = 1 line, i.e. cell of type VTK_LINE), without loss of data (arrays are passed to output). Note that conversion into molecule requires a one-dimensional array to be used as Atomic Numbers.
These conversion filters allow all polydata filters to be used for molecule analysis, as shown on figure 1.
Parallel Molecule Support
The molecule framework has been improved to support distributed parallelism with MPI. Now vtkMolecule and vtkMoleculeMapper support and manage ghost atoms and ghost bonds allowing the distribution of big molecules (or a set of atoms) on multiple processors.
The subdivision of a dataset into pieces can be done through the recently created “Distribute Points” filter (vtkDistributedPointCloudFilter): it creates spatially contiguous region, containing an equivalent number of points (see figure 2b).
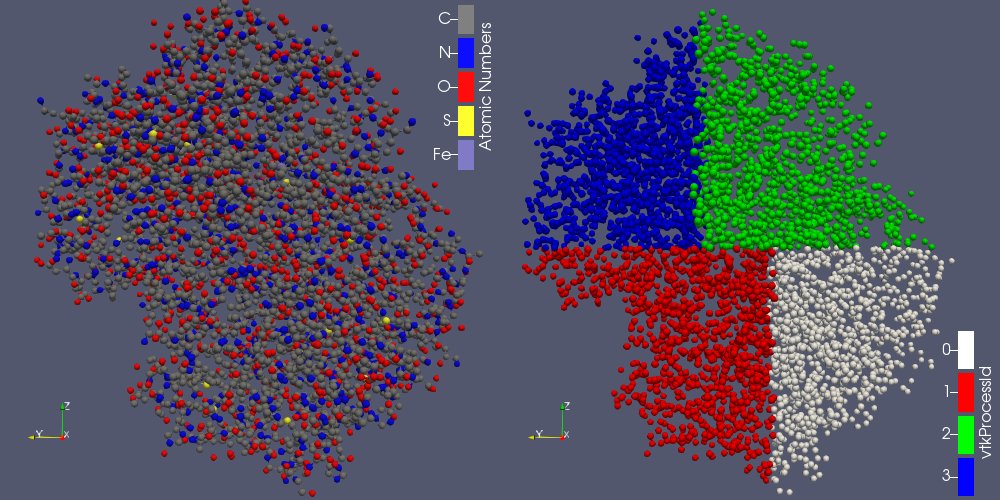
In ParaView, when the rendering is done on the client-side, the different pieces have to be merged before being rendered. It is possible thanks to the Append Molecule filter (vtkMoleculeAppend)
Bonds Computation
A VTK filter (vtkSimpleBondPerceiver) aims to create bonds in a molecule, depending on the inter-atomic distance between and the atoms Van Der Waals radius. This filter was improved in two ways:
- As only the nearest neighbours of an atom can have a bond with it, the main algorithm now extracts a list of good candidates for bonds (thanks to a locator), reducing the time of computation.
- The new filter vtkPSimpleBondPerceiver extends the original filter for molecules distributed over multiple MPI ranks (it will detect and create bonds between atoms located in neighbor ranks and create ‘ghost’ atoms accordingly). This feature enables processing large atoms sets and gives better performance.
You can now use this filter in ParaView under the name “Compute Bonds” (see Figure 3).
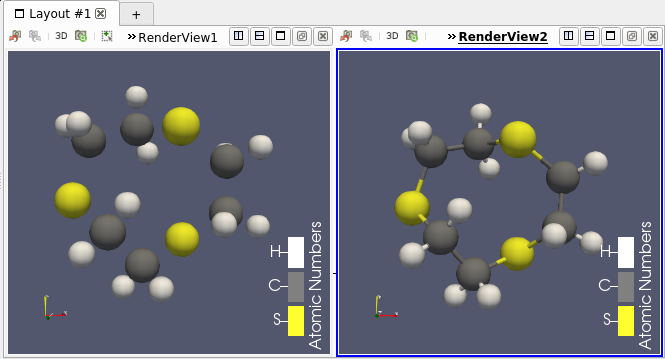
Support for Molecule in Programmable filter/source
This molecule framework can be extended with python and in the Programmable filter/source module, as they now support molecule as input and output. Molecule can be easily generated and processed (see Figures 4 and 5)
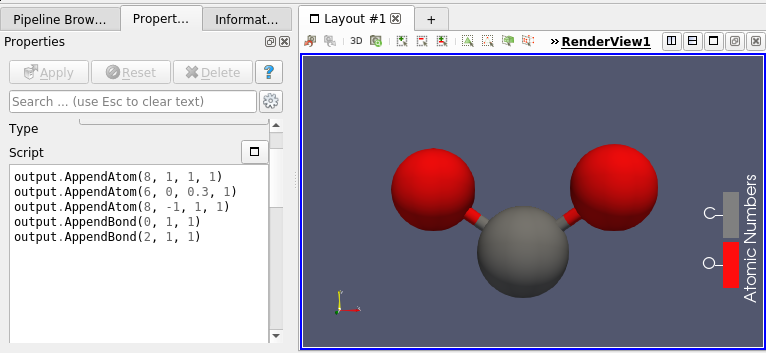
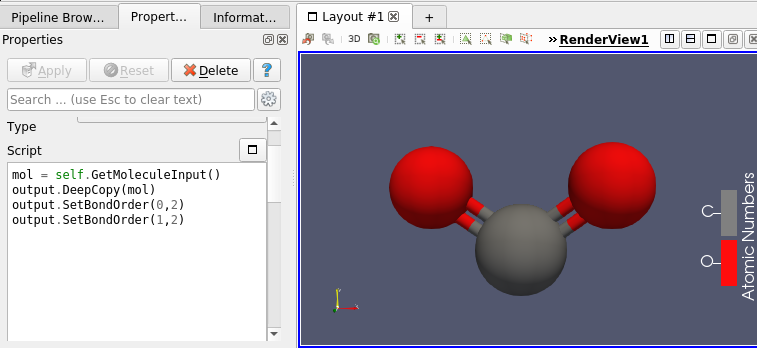
Going Further
For more information about VTK/ParaView molecule framework, please refer to these previous blog entries:
https://blog.kitware.com/new-features-paraview-chemistry-pettt/
https://blog.kitware.com/rendering-engine-improvements-in-vtk/
Acknowledgements
This work is a result of a common effort by Kitware France and the CEA (French Alternative Energies and Atomic Energy Commission) in the context of the project SMICE – a collaborative research project about material exaflopic simulation founded by the French program “Investissements d’Avenir”.
 |
 |
Really nice filters and blog post, Nicolas and company! It is great to see these advances in molecular data support in ParaView.
This addition might be very beneficial for my soft-body granular dynamics simulations where particles consist of spheres. Excited to try it out!
Great work, and nice post describing the additions. It is great to see the support for distributed molecule data sets being developed. Did you measure any benchmarks on really large systems that you can share? It looks like this is aimed at supporting some pretty large temporal atomic trajectories, and it should enable scaling to beyond the millions of atoms supported by rendering on a single node.